材料研发中传统“试错法”依赖大量重复实验,严重制约了新材料的研发进程。理论模拟能够建立材料的结构‑物性关联,显著加速材料研发速度。常用的密度泛函理论(DFT)非常精确,但无法满足模拟规模需求;经验力场可以大规模模拟,但其精度差,难以描述复杂体系演化。近年来发展的机器学习力场有望实现可比DFT精度、大规模和长时间的模拟。
近期,大连理工大学高峻峰教授、南京大学张荣院士、王学锋教授、中国科学院大学周武教授和中国科学院金属研究所杨腾研究员等多个科研单位合作,在PdTe材料合成机理上取得了重要突破。PdTe是近期备受关注的拓扑狄拉克半金属。目前基于电化学处理等PdTe制备方法,难以避免PdTe2杂相产生,大面积、厚度可控的PdTe材料合成仍面临着显著挑战。王雪峰教授等不仅实现大面积、均匀PdTe薄膜的制备,还能够可控合成PdTe2/PdTe异质界面,具有高效手性太赫兹发射性能。成果以“Large-area non-stoichiometric phase transition in transition metal chalcogenide films”为题并于2026年1月发表在《Nature Materials》期刊 [DOI:10.1038/s41563-025-02471-9]。
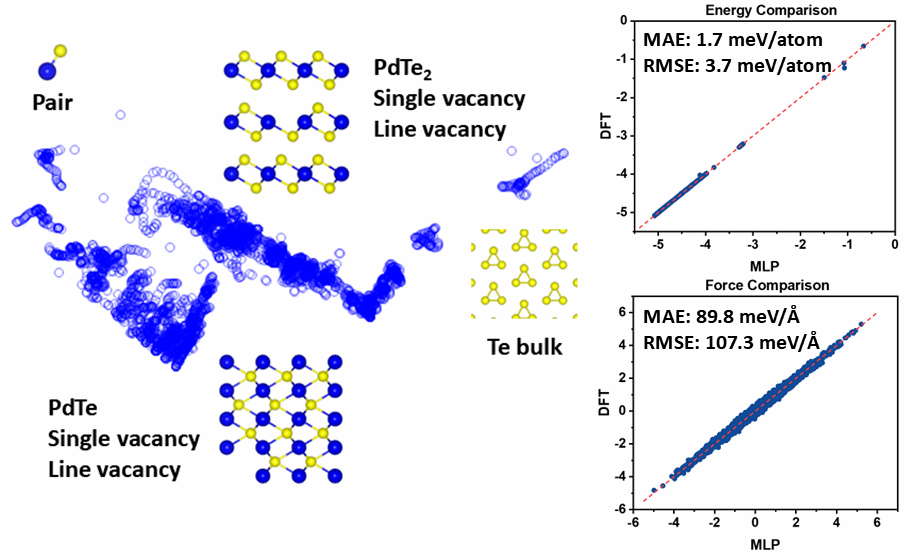
图1. 机器学习数据集组成,机器学习力场与DFT精度对比
问鼎网中国高峻峰教授长期从事材料结构重构、生长机理及界面催化的动力学研究,团队构建了可与DFT媲美且支持大规模模拟的机器学习力场,并发展了主动学习工作流策略。在新材料预测、材料生长和相变、表界面重构演化、催化等复杂材料研究体系做出了系列突出性成果。
在本工作中,高峻峰教授和常远博士后发展了包含Pd、Te单质以及含缺陷的PdTe2和PdTe等多种小尺度模型,采用密度泛函理论(DFT)计算,生成了约21.8万组结构‑能量‑力的数据集(图1)。基于此数据集训练得到的机器学习力场,在保持与DFT相当精度的同时,实现大尺度、长时间的模拟,采用机器学习力场,揭示了PdTe2到PdTe热相变的关键动力学机理。
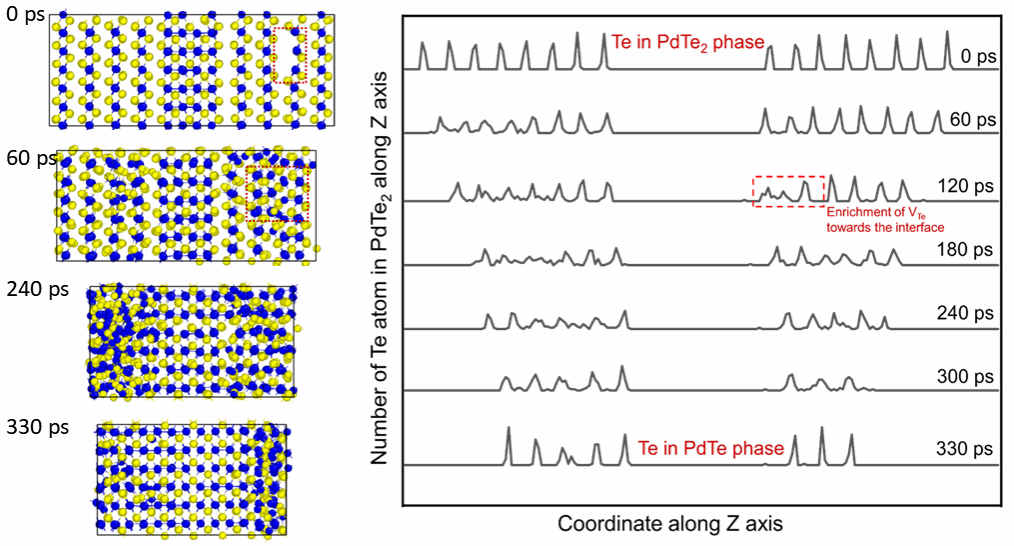
图2. PdTe2到PdTe的连续相变模拟和Te缺陷向PdTe2/PdTe界面的聚集
PdTe2到PdTe的相变的机器学习分子动力学模拟指出相变优先在缺陷附近触发,并呈现出由下至上、逐层推进的转变模式。通过对体系中Te原子数目的统计分析,发现Te空位产生后倾向于向PdTe2/PdTe界面处富集(图2)。该现象可通过过渡态计算进一步阐明:PdTe2中Pd原子与Te空位的扩散势垒均极低,因而热处理引入的Te空位得以在层间迁移,同时Pd原子也更易进入范德华间隙,从而驱动相变进行(图3)。
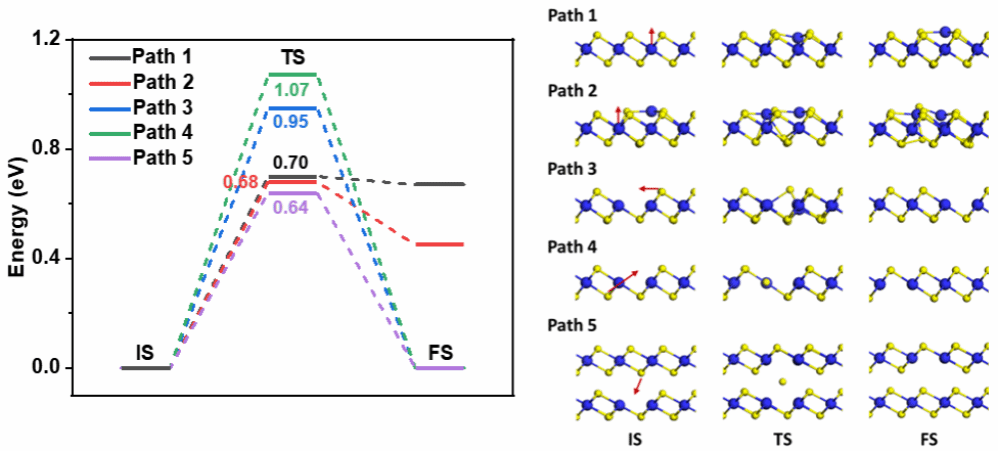
图3. PdTe2中Pd原子和Te空位的扩散路径和势垒
团队还进一步发展了PtTe2/PtTe的机器学习力场,植入分子动力学后,长时间模拟发现PtTe2也存在向PtTe热相变,其过程与PdTe2到PdTe的热相变类似,从而推理类似结构的过渡金属碲化物的热相变具有一定的普适性机理。
南京大学博士后陈中强、中国科学院大学时金安、中国科学院金属研究所黄建啟、大连理工大学博士后常远为本文共同第一作者。中国科学院大学周武教授、中国科学院金属研究所杨腾研究员、大连理工大学高峻峰教授、南京大学张荣教授和王学锋教授为共同通讯作者。
📄论文信息:Nature Materials, DOI: 10.1038/s41563-025-02471-9
🔗全文链接:http://www.nature.com/articles/s41563-025-02471-9

